Figure
1.
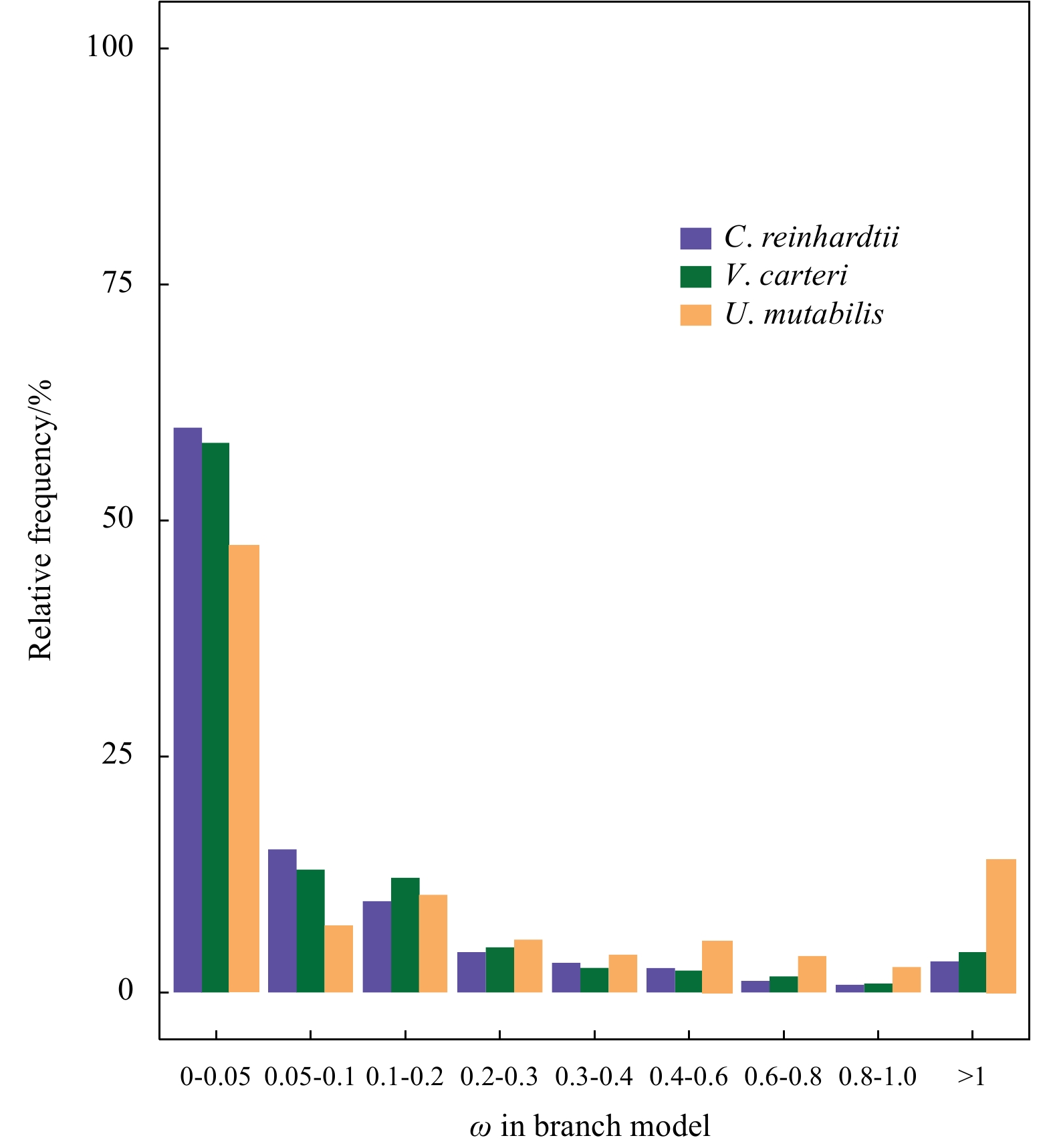
Comparision of dN/dS among U. mutabilis, C. reinhardtii and V. carteri. Significance of the deviations was calculated by using Wilcoxon rank sum test.
| Citation: | Jian Zhang, Xiaowen Zhang, Wentao Han, Xiao Fan, Yitao Wang, Dong Xu, Yan Zhang, Jian Ma, Chengwei Liang, Naihao Ye. Identification of genes under positive selection reveals evolutionary adaptation of Ulva mutabilis[J]. Acta Oceanologica Sinica, 2020, 39(10): 35-41. doi: 10.1007/s13131-020-1658-1

|
Green algae especially ulvophytes are attractive model systems for understanding growth, development, and evolution (Cocquyt et al., 2010), and are key to understand the evolution of multicellularity in the green lineage (Wichard et al., 2015). These algae are also key contributors to coastal biogeochemical cycles, especially to the marine sulfur cycles, because they produces high levels of dimethylsulfoniopropionate, the main precursor of volatile dimethyl sulfide (Van Alstyne, 2008). Their rapid and abundant growth makes them untapped resources for food, fuel, and high-value compounds, but they also lead to significant environmental consequences in the form of green tides and biofouling (Vesty et al., 2015; Smetacek and Zingone, 2013). In recent years, green tides have received increasing attention because of well-publicized blooms in China and France. Massive green tides caused mainly by Ulva prolifera have occurred successively for 13 years (2007–2019) in the Yellow Sea coastal region of China (Zhang et al., 2019). Blooms of Ulva species have occurred in Brittany, France since the 1980s where they accumulate to depths of up to one meter (Charlier et al., 2006).
Unlike land plants and unicellular green algae, mechanism studies of growth and development at the molecular level in multicellular green seaweeds are currently very limited. Until now, only one Ulva genome, that of Ulva mutabilis, has been sequenced. Ulva mutabilis is a ubiquitous representative of class Ulvophyceae (De Clerck et al., 2018). The U. mutabilis genome sequence provides opportunities to understand the fundamental evolution of the Ulva green lineage.
Detection of genes or genomic regions that have been targeted by positive selection can help to understand the processes of evolution and adaptation (Jensen and Bachtrog, 2010). In this study, we performed a genome-wide analysis to detect genes under positive selection (GUPS) in U. mutabilis. We used single-copy orthologous families (n=3 905) present in U. mutabilis, Chlamydomonas reinhardtii, and Volvox carteri. Chlamydomonas reinhardtii and V. carteri were used as out groups to identify signatures of positive selection in U. mutabilis. Our results shed light on the adaptive evolution of functional genes in Ulva species and revealed how they have diverged to thrive under various environmental conditions.
To explore the role of positive selection in the adaptive patterns of U. mutabilis, protein-coding sequences were downloaded from the website https://bioinformatics.psb.ugent.be/orcae/overview/Ulvmu. We chose C. reinhardtii and V. carteri as the out groups and their coding sequences were acquired from JGI. We selected v5.6 version of C. reinhardtii and v2.1 of V. carteri among various versions.
Furthermore, to define a set of conserved genes for cross-taxa comparison, we employed Orthofinder (v2.3.3) to search homologous genes of three species based on nucleotide sequence (Emms and Kelly, 2015). The lengths under 150 bp of sequences were discarded and stop codons were removed from the sequences prior to alignment.
Alignment of these proteins was performed using mafft (v7) (Nakamura et al., 2018). Codon alignments were generated using the protein sequence alignments as a guide by PAL2NAL (Suyama et al., 2006). All gaps in alignment were cut off in order to alleviate the effect of ambiguous bases on the inference of positive selection, and all sequence alignment results were saved as PAML format (Suyama et al., 2006).
The ratio of non-synonymous (dN) to synonymous (dS) nucleotide substitutions (dN/dS) ω provides information about the evolutionary forces operating on a gene (Biswas and Akey, 2006). If there is no environmental pressure, gene are in neutral selection by an ω=1. If dN is beneficial for organisms, genes are under positive selection which ω>1. On the contrary, genes are in purifying selection with ω<1 (Yang, 2007).
Firstly, to calculate specific branch of each gene family in the three species’s evolutionary rates, the codeml program in the PAML (v4.9) package with the free-ratio model (M=1) was operated on each orthogroups (Yang, 2007). The user tree was assumed to be [(U. mutabilis), (C. reinhardtii, V. carteri)] for all genes. We filtered dS>3 or dN/dS>3 to eliminate the effect of outliers. Significance of the deviations from the median dN/dS ratio between three species branches were detected using Wilcoxon rank sum test. As free-ratio model calculates the values of different branches without test, we then used branch model (M=2) of Codeml program in the PAML package to calculate ω of the foreground branch U. mutabilis. The null model (M=0), in which one ω value was assumed for all branches, was used for likelihood ratio test (LRT) to identify genes of ω>1.
However, for single copy genes, most of codon sites in the branch are supposed to be highly conserved to maintain protein function (Swift et al., 2016). So there must be a lot of sites that are less than 1. Therefore, we attempt to determine positive selection sites in each gene. We then used site-specific model which assumes that selection pattern varies among sites in the alignment but not among branches in the phylogeny. We used a pair of site model comparisons to test for positive selection (M7 vs. M8). LRT was performed to test which model fits the data best. We used chi-square test with the degrees of freedom of two to calculate twice the difference in log-likelihood values between the models. Using the p.adjust function in fdrtool R package, the FDR correction was applied to the P values with a significance level of 0.05 (Bakewell et al., 2007; R Development Core Team, 2014).
Finally, to find positive selection evidence of specific sites in specific lineage, the improved branch-site model A (model=2, Nsites=2, fixed omega=0, omega=2) and null model (model=2, Nsites=2, fixed omega=1, omega=1) was used, which was proven to be more sensitive than branch model or site model (Yang and Reis, 2011). We selected the U. mutabilis branch as the foreground branch with the C. reinhardtii and V. carteri as background branches. All gaps in alignment were cut off in order to alleviate the effect of ambiguous bases on the inference of positive selection. Each single-copy gene family runs both model A and null models. Then based on the results of the two models, we used likelihood ratio test (LRT) with a chi square distribution in one degree of freedom to determine whether there are positive selections at a threshold of P<0.05. If model A fits adapts the data, then we used the paml data to find out whether there are positive selection sites and sites was significant or not.
To identify the physiological processes involved by Genes Under Positive Selection of U. mutabilis, NCBI non-redundant protein (Nr), Protein family (Pfam) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways annotation was performed. The website https://www.genome.jp/tools/kaas/ was used to find KEGG pathways, and the KOBAS (v3.0) (Xie et al., 2011) was used to test the statistical enrichment of PSGs in KEGG pathways (Kanehisa and Goto, 2000).
We found 5 252 homologous gene families in the genomes of U. mutabilis, C. reinhardtii, and V. carteri, and among them, 3 925 were single-copy homologous gene families. After discarding sequences <150 bp in length, the remaining sequences (n=3 905) were analyzed further. There were also 1 336 amplified gene families and 120, 410, and 482 species-specific expansion homologous gene families in U. mutabilis, C. reinhardtii, and V. carteri, respectively.
We constructed a species phylogenetic tree and used it for the positive selection analysis of each single-copy homologous gene families. Under the branch model, we found that the ratio of non-synonymous (dN) to synonymous (dS) changes (dN/dS ratio) was mainly in the range 0–0.2 in all three species, suggesting strong purifying selection for the single-copy genes (Fig. 1). The median of the dN/dS ratio in U. mutabilis (0.378) was significantly higher than that in the other two species (0.127 and 0.161) (Fig. 1). The frequency distribution of dN/dS ratios clearly showed that U. mutabilis had more genes with high dN/dS ratios (dN/dS>0.4) than the other species (Fig. 2). We also compared the two-ratio and one-ratio models using the likelihood ratio test (LRT) and found that nine GUPS in U. mutabilis genes (Table 1).

| Comparison | U. mutabilis | C. reinhardtii | V. carteri |
| Branch model | |||
| Free-radio model | |||
| Mean ω | 0.378 | 0.127 | 0.161 |
| Two-radio model | |||
| Number of GUPS (1<ω<3) | 9 | ||
| Site model | |||
| Number of GUPS | 242 | ||
| Number of GUPS (FDR<0.05) | 237 | ||
| Number of GUPS (PP>0.9) | 30 | ||
| Branch-site model | |||
| Number of GUPS | 67 | ||
| Number of GUPS (FDR<0.05) | 63 | ||
| Number of GUPS (PP>0.9) | 46 |
 DownLoad:
CSV
DownLoad:
CSV
The random-site model, which ignores ω variation among lineages, was used to identify sites in genes that were targets of positive selection. After the LRT analysis, we detected 242 orthologous GUPS. Then we used a false discovery rate (FDR) of 5% to exclude false positive selection, and finally obtained 236 candidate GUPS and 30 of them were prominent (posterior probability (PP) >0.9). We used KEGG pathways to annotate the genes and 53 of them were assigned to pathways. Three pathways were highly enriched, namely ribosome in genetic information processing (p=0.002), photosynthesis-antenna proteins in energy metabolism (p=0.01), and phagosome in transport and catabolism (p=0.045).
Finally, we used the branch-site model to detect evidence of positive selection in U. mutabilis. A total of 67 GUPS were identified with chi-square distribution values higher than the critical value of 3.84. After the FDR correction, 63 genes were found to be significant. Then the Bayes empirical Bayes (BEB) approach was applied to calculate the posterior probabilities (PP) to identify significant GUPS with p<0.05 and PP>0.9, and a total of 46 genes were selected. Further, we determined proprietary positive selection sites and found 627 (PP>0.9) and 150 (PP>0.99) positively selected sites in the 46 genes. The distribution of KEGG classification of the 63 GUPS showed that three categories of pathways were common for all the genes (Table 2). Among them, metabolic processes were the most enriched, including amino acid metabolism (3), energy metabolism (2, photosynthesis), metabolism of cofactors and vitamins (2, ubiquinone and riboflavin biosynthesis), nucleotide metabolism (2, purine metabolism), metabolism of terpenoids and polyketides (2, chlorophyll an+++d carotenoid biosynthesis), carbohydrate metabolism (1), and lipid metabolism (1), followed by genetic information processing with nine GUPS that were mainly involved in ribosome biogenesis, translation, and folding. The third most enriched category had six GUPS that were mainly involved in environmental information processing (phosphatidylinositol signaling system and MAPK signaling pathway), signaling and cellular processes (chromosome and cytoskeleton proteins), and mineral absorption (copper transporter).
| Protein ID | χ2 | p-value | Nr | KEGG |
| UM051_0030.1 | 3.649032 | 0.049775 | photosystem I reaction center subunit VI-chloroplastic-like | photosynthesis |
| UM041_0034.1 | 3.866748 | 0.049252 | 20S proteasome beta subunit | proteasome |
| UM025_0090.1 | 3.875197 | 0.049005 | transcription factor Tfb4 | basal transcription factors |
| UM020_0175.1 | 3.8908 | 0.048551 | cyclophilin-like protein | |
| UM017_0023.1 | 3.90656 | 0.048098 | type I inositol polyphosphate 5-phosphatase 1-like isoform X1 | |
| UM061_0055.1 | 3.92971 | 0.04744 | spermatogenesis-associated protein 4 | |
| UM020_0022.1 | 3.943346 | 0.047057 | MATE efflux family | |
| UM011_0045.1 | 3.952812 | 0.046793 | adenosine/AMP deaminase family protein | metabolic pathways |
| UM015_0094.1 | 3.96594 | 0.04643 | SET domain-containing protein | |
| UM002_0196.1 | 3.986382 | 0.045869 | tetratricopeptide repeat protein | |
| UM059_0039.1 | 3.988128 | 0.045822 | SET domain-containing protein | |
| UM005_0194.1 | 4.029512 | 0.044711 | metallo-hydrolase oxidoreductase | |
| UM119_0020.1 | 4.035582 | 0.04455 | dynein light chain, type 1 | |
| UM110_0011.1 | 4.076886 | 0.043474 | riboflavin biosynthesis chloroplastic | riboflavin metabolism |
| UM009_0042.1 | 4.089456 | 0.043152 | flavo protein | |
| UM119_0008.1 | 4.096726 | 0.042966 | indole-3-glycerol-phosphate synthase | |
| UM001_0277.1 | 4.121502 | 0.042341 | la-related protein 1A-like | |
| UM003_0103.1 | 4.175942 | 0.041002 | chlorophyll a-b binding protein of LHCII | photosynthesis-antenna protein |
| UM075_0040.1 | 4.193048 | 0.04059 | Sac domain-containing phosphoinositide phosphatase | |
| UM020_0063.1 | 4.271742 | 0.038751 | 50S ribosomal protein L3-1, chloroplastic | ribosome |
| UM101_0006.1 | 4.318366 | 0.037703 | tubulin-tyrosine ligase | |
| UM002_0307.1 | 4.357032 | 0.036856 | nucleotide-diphospho-sugar transferase domain | |
| UM033_0058.1 | 4.399316 | 0.035953 | vacuolar fusion protein MON1 homolog isoform X2 | |
| UM022_0088.1 | 4.428682 | 0.03534 | DUF455 family | |
| UM004_0158.1 | 4.448474 | 0.034932 | integral membrane protein TerC, riboswitch-linked | |
| UM001_0470.1 | 4.459038 | 0.034717 | WD repeat-containing protein 6 isoform X1 | |
| UM004_0249.1 | 4.51936 | 0.033513 | ABC transporter F family member-like | |
| UM095_0035.1 | 4.519716 | 0.033506 | ubiquinone biosynthesis protein, partial | |
| UM019_0140.1 | 4.552578 | 0.032869 | 40S ribosomal protein S8 | ribosome |
| UM100_0037.1 | 4.616152 | 0.031672 | hypothetical protein | |
| UM133_0013.1 | 4.619028 | 0.031619 | Reticulata-related chloroplastic-like | |
| UM077_0057.1 | 4.643308 | 0.031175 | IMPACT isoform X1 | |
| UM005_0088.1 | 4.64428 | 0.031157 | thylakoid lumenal protein | |
| UM005_0011.1 | 4.647732 | 0.031095 | RNA polymerase II-associated factor 1-like protein | |
| UM007_0229.1 | 4.671324 | 0.03067 | sorting nexin 2a | |
| UM035_0106.1 | 4.861868 | 0.027457 | ribosome 60S biogenesis N-terminal-domain-containing protein | |
| UM098_0047.1 | 4.880034 | 0.027169 | phospholipase A I-like isoform X2 | |
| UM040_0040.1 | 4.933432 | 0.026342 | CUE domain-containing protein | |
| UM057_0023.1 | 5.000268 | 0.025343 | COMPASS-like H3K4 histone methylase component WDR5A | |
| UM085_0051.1 | 5.080884 | 0.024191 | GPI inositol-deacylase PGAP1-like isoform B | |
| UM062_0033.1 | 5.242224 | 0.022045 | argininosuccinate synthase | alanine, aspartate metabolism |
| UM014_0155.1 | 5.399928 | 0.020138 | polysulfide reductase | |
| UM066_0033.1 | 5.46381 | 0.019414 | epsilon-COP | |
| UM020_0154.1 | 5.538692 | 0.0186 | transcription factor bHLH34 | |
| UM066_0060.1 | 5.542258 | 0.018563 | L-isoaspartate(D-aspartate) O-methyltransferase | |
| UM041_0094.1 | 5.61881 | 0.017769 | prephenate dehydratase | biosynthesis of amino acids |
| UM051_0040.1 | 5.620024 | 0.017756 | centrosomal protein of 78 kDa | |
| UM047_0010.1 | 5.762886 | 0.016368 | phosphatidate phosphatase PAH1 isoform X1 | glycerophospholipid metabolism |
| UM002_0428.1 | 5.880034 | 0.015314 | PREDICTED: nuclear-interacting partner of ALK isoform X1 | |
| UM020_0144.1 | 6.001238 | 0.014296 | kinesin light chain 3 isoform X1 | |
| UM028_0126.1 | 6.071472 | 0.013738 | glycine cleavage system H protein, mitochondrial | glycine, serine and threonine metabolism |
| UM023_0033.1 | 6.119312 | 0.013371 | adenylyl cyclase class-3/4/guanylyl cyclase | purine metabolism |
| to be continued | ||||
DownLoad:
CSV
Besides the single-copy gene families, we also conducted a positive selection analysis of the 120 U. mutabilis-specific amplified gene families (Table 3). Only two of these gene families were identified as under positive selection under the branch model, whereas 37 and 13 gene families were found under positive selection using the site and branch-site models respectively. These genes were annotated with KEGG pathways, including biosynthesis of amino acids, carbon fixation in photosynthesis, ubiquitin mediated proteolysis, peroxisome, pyrimidine metabolism, spliceosome, protein export, and protein processing in endoplasm. The specific function of these amplified gene families was listed by searching Nr and Pfam databases.
| Model | Gene ID | Function description in Nr and Pfam database | Gene ID | Function description in Nr and Pfam database |
| Branch-model | UM011_0230.1 | p25-alpha | UM068_0038.1 | polyketide cyclase/dehydrase and lipid transport |
| UM011_0231.1 | UM094_0042.1 | |||
| Site-model | UM031_0027.1 | glucokinase | UM005_0209.1 | aminotransferase class I and II |
| UM031_0028.1 | UM005_0214.1 | |||
| UM146_0032.1 | N2, N2-dimethylguanosine tRNA methyltranse | UM008_0174.1 | hypothetical protein | |
| UM146_0033.1 | UM281_0004.1 | |||
| UM013_0057.1 | Dor1-like family | UM100_0006.1 | chloroplastic isoform | |
| UM060_0118.1 | UM100_0007.1 | |||
| UM049_0058.1 | ubiquitin-conjugating enzyme | UM018_0136.1 | peptidase family M41 | |
| UM058_0003.1 | UM020_0045.1 | |||
| UM012_0017.1 | protein kinase domain | UM015_0045.1 | protein tyrosine kinase | |
| UM149_0036.1 | UM015_0046.1 | |||
| UM093_0008.1 | recA bacterial DNA recombination protein | UM001_0125.1 | ubiquitin-specific protease | |
| UM093_0026.1 | UM001_0129.1 | |||
| UM005_0351.1 | cytochrome C biogenesis protein | UM001_0491.1 | FAD dependent oxidoreductase | |
| UM077_0032.1 | UM001_0492.1 | |||
| UM010_0035.1 | TCP-1/cpn60 chaperonin | UM002_0402.1 | DNL zinc finger | |
| UM011_0180.1 | UM018_0127.1 | |||
| UM011_0077.1 | hypothetical protein | UM037_0017.1 | protein of unknown function (DUF3250) | |
| UM011_0096.1 | UM037_0018.1 | |||
| UM037_0056.1 | no hit | UM034_0001.1 | FKBP-type peptidyl-prolyl cis-trans isomerase | |
| UM044_0086.1 | UM034_0003.1 | |||
| UM009_0050.1 | CobW/HypB/UreG, nucleotide-binding domain | UM008_0173.1 | WD domain, G-beta repeat | |
| UM092_0039.1 | UM281_0003.1 | |||
| UM069_0030.1 | carbamoyl-phosphate synthase small chain, CPSase domain | UM015_0020.1 | cation efflux family | |
| UM309_0004.1 | UM026_0097.1 | |||
| UM043_0048.1 | aminotransferase class I and II | UM012_0077.1 | no hit | |
| UM057_0008.1 | UM131_0006.1 | |||
| UM001_0588.1 | plasma-membrane choline transporter | UM001_0573.1 | Sec63 Brl domain | |
| UM001_0591.1 | UM002_0245.1 | |||
| UM035_0019.1 | TIP41-like family | UM003_0004.1 | ATP12 chaperone protein | |
| UM035_0020.1 | UM047_0029.1 | |||
| UM010_0149.1 | TspO/MBR | UM007_0020.1 | ABC transporter | |
| UM010_0150.1 | UM139_0019.1 | |||
| UM012_0035.1 | Hsp70 protein | UM008_0176.1 | enoyl-(acyl carrier protein) reductase | |
| UM012_0036.1 | UM281_0001.1 | |||
| UM018_0002.1 | RNA methyltransferase | UM103_0009.1 | no hit | |
| to be continued | ||||
DownLoad:
CSV
Orthologs are genes that have evolved from a common ancestral gene via speciation. To investigate the selective pressures at the branch level in U. mutabilis and related species, we estimated the substitution rates for each orthogroup. The median of the dN/dS ratio in U. mutabilis was significantly larger than that in C. reinhardtii and V. carteri, which strongly supported the accelerated evolution of U. mutabilis after splitting from its ancestral lineage (Fig. 1). The accelerated evolution of genes is often driven by positive selection or relaxed selection pressure. Green macroalgae mostly belong to class Ulvophyceae, the main multicellular branch of class Chlorophyceae, and constitute important primary producers of coastal ecosystems (Wichard et al., 2015). Fluctuating environmental conditions, characterized by intense stresses such as extreme temperatures, rapid salinity and nutrient changes, desiccation, and intense sunlight, are major inducers in the evolution of intertidal macroalgae (Kakinuma et al., 2006). We speculated that the high evolutionary rate in U. mutabilis is due mainly to positive selection rather than relaxed selection pressure.
Photosynthesis genes have been fine-tuned over billions of years as a result of natural selection (Niinemets et al., 2017). Two genes related to the photosynthetic apparatus were identified to be under adaptive evolution, supporting the idea that Ulva species may have evolved to maintain photosynthetic efficiency under tidal environments. The thylakoid membrane-integral light-harvesting complex (LHC) antenna systems, which are encoded by a multigene family of LHC genes, play important roles in regulating energy flow to photosynthetic reaction centers (Neilson and Durnford, 2010). The LHC systems harvest and transfer excitation energy to drive photosynthesis. However, under excess light conditions, they undergo a conformational change and activate a quenching state to dissipate energy in order to protect the photosystem. In our analysis, an LHCII gene, encoding a light harvesting protein in photosystem II, was found to be under adaptive evolution in U. mutabilis. Evidence of adaptive evolution in U. mutabilis photosynthetic apparatus also was found in photosystem I reaction center subunit VI. This result is in accordance with a previous study that found that the Ulva photosystem I had higher tolerance to osmotic stress than photosystem II, and that PSI-driven cyclic electron flow allowed Ulva species to survive in desiccated conditions (Gao et al., 2014, 2011, 2015).
Signatures of adaptive evolution were identified in antioxidant systems, including xanthophyll cycle (Xc) and photorespiration. The Xc involves violaxanthin de-epoxidase (VDE) and the zeaxanthin epoxidase (ZEP) and is one of the most rapid and efficient photoprotection mechanisms of plant and algae to high irradiance (Zhang et al., 2015; Xie et al., 2013). The photoprotection mechanism of non-photochemical quenching in Ulva linza was shown to be controlled to a great extent by Xc, which is more similarity to the mechanism in Arabidopsis than to that in Chlamydomonas (Zhang et al., 2015). In addition, VDE and ZEP were found to be permanently operating to maintain the dynamic between lipid and LHCII subunits under moderate light conditions in Ulva species (Xie et al., 2013). The retained Xc pigments regulated the fluidity of the thylakoid membrane, protected the thylakoid membrane from oxidative damage, and reduced potential production of reactive oxygen species (ROS) by consuming oxygen that is introduced into zeaxanthin by ZEP (Xie et al., 2013). The permanent cycling of Xc pigments in the regulation of membrane fluidity and reduction of the dioxygen level was found to be important for Ulva survival under both excess light and desiccation (Gao et al., 2015). The adaptive evolution of the ZEP gene in U. mutabilis found in our analysis further confirmed the essential function of Xc for the successful colonization in coastal ecosystems by Ulva species.
Photorespiration is an important mechanism that protects cells from photooxidative damage by regulating energy demand and oxygen consumption (Wingler et al., 2000). In addition, photorespiratory glycine facilitates the accumulation of glutathione to protect the photosynthetic components (Noctor et al., 1999). We found one gene encoding mitochondrial glycine cleavage system H protein that participates in photorespiration was under adaptive evolution. This result indicates that photorespiration may be enhanced in Ulva species to minimize production of ROS in the chloroplasts and mitigate oxidative damage under costal stress conditions.
Ulva species are known for their rapid growth, proliferation, and phenotypic plasticity. In our study, evidence of positive selection was found in genes associated with chlorophyll, purine, cellulose, amino acid, and protein biosynthesis processes that may be related to the proliferation of Ulva species. Besides the light harvesting LHCII, the gene encoding geranylgeranyl reductase, which is involved in chlorophyll synthesis, was under positive selection in U. mutabilis. Both these two genes play essential roles in photosynthesis and therefore growth. However, fast growth can be achieved only if the photosynthetic production of ATP, NADPH, and organic carbon is in balance with anabolism (Teng et al., 2017). The presence of GUPS associated with nucleic acid, protein, and cell wall polysaccharide biosynthesis suggested that selection also affected the speed at which photosynthetic products were transformed into biomass. Genes encoding adenosine deaminase and adenylyl cyclase class-3/4/guanylyl cyclase participate in purine metabolism and the latter also can generate cGMP, which is an important secondary messenger in signal transduction systems. Besides, the GUPS encoding RNA polymerase II-associated factor and La-related protein participate in RNA synthesis. Among these genes, we detected a gene that encodes the nucleotide-diphospho-sugar transferase domain, which is the catalytic subunit of cellulose synthase that functions in cell wall synthesis. The signatures of adaptive evolution were found in several genes involved in rRNA processing (ribosomal proteins), translation (transcription factors, tubulin-tyrosine ligase), folding (cyclophilin), and transport (clathrin light chain, vacuolar fusion protein, sorting nexin), indicating that adaptive evolution was associated with the regulation of protein synthesis. Ribosomes are essential for protein synthesis in all living cells and play a distinct role in photosynthesis, plant development, and stress tolerance (Zhang et al., 2016).
Inositol phospholipids have long been known to have an important regulatory role in cell physiology. Besides classical signal transduction at the cell surface, they also regulate membrane traffic, the cytoskeleton, nuclear events, and the permeability and transport functions of membranes (Di Paolo and De Camilli, 2006). Three genes encoding inositol polyphosphate 5-phosphatase, phospholipase A, and phosphoinositide phosphatase, which participate in the phosphatidylinositol signaling system, were found to be under adaptive evolution in U. mutabilis. We propose that the phosphatidylinositol signaling system may play important roles in the stress adaptation, complex morphology formation, and rapid growth of Ulva species.
| [1] |
Bakewell M A, Shi Peng, Zhang Jianzhi. 2007. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proceedings of the National Academy of Sciences of the United States of America, 104(18): 7489–7494. doi: 10.1073/pnas.0701705104
|
| [2] |
Biswas S, Akey J M. 2006. Genomic insights into positive selection. Trends in Genetics, 22(8): 437–446. doi: 10.1016/j.tig.2006.06.005
|
| [3] |
Charlier R H, Morand P, Finkl C W, et al. 2006. Green tides on the Brittany coasts. In: 2006 IEEE US/EU Baltic International Symposium. Klaipeda, Lithuania: IEEE, 1–13
|
| [4] |
Cocquyt E, Verbruggen H, Leliaert F, et al. 2010. Evolution and cytological diversification of the green seaweeds (Ulvophyceae). Molecular Biology and Evolution, 27(9): 2052–2061. doi: 10.1093/molbev/msq091
|
| [5] |
De Clerck O, Kao S M, Bogaert K A, et al. 2018. Insights into the evolution of multicellularity from the sea lettuce genome. Current Biology, 28(18): 2921–2933. doi: 10.1016/j.cub.2018.08.015
|
| [6] |
Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature, 443(7112): 651–657. doi: 10.1038/nature05185
|
| [7] |
Emms D M, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology, 16(1): 157. doi: 10.1186/s13059-015-0721-2
|
| [8] |
Gao Shan, Gu Wenhui, Xiong Qian, et al. 2015. Desiccation enhances phosphorylation of PSⅡ and affects the distribution of protein complexes in the thylakoid membrane. Physiologia Plantarum, 153(3): 492–502. doi: 10.1111/ppl.12258
|
| [9] |
Gao Shan, Shen Songdong, Wang Guangce, et al. 2011. PSI-driven cyclic electron flow allows intertidal macro-algae Ulva sp. (Chlorophyta) to survive in desiccated conditions. Plant and Cell Physiology, 52(5): 885–893. doi: 10.1093/pcp/pcr038
|
| [10] |
Gao Shan, Zheng Zhenbing, Gu Wenhui, et al. 2014. Photosystem I shows a higher tolerance to sorbitol-induced osmotic stress than photosystem Ⅱ in the intertidal macro-algae Ulva prolifera (Chlorophyta). Physiologia Plantarum, 152(2): 380–388. doi: 10.1111/ppl.12188
|
| [11] |
Jensen J D, Bachtrog D. 2010. Characterizing recurrent positive selection at fast-evolving genes in Drosophila miranda and Drosophila pseudoobscura. Genome Biology and Evolution, 2: 371–378. doi: 10.1093/gbe/evq028
|
| [12] |
Kakinuma M, Coury D A, Kuno Y, et al. 2006. Physiological and biochemical responses to thermal and salinity stresses in a sterile mutant of Ulva pertusa (Ulvales, Chlorophyta). Marine Biology, 149(1): 97–106. doi: 10.1007/s00227-005-0215-y
|
| [13] |
Kanehisa M, Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 28(1): 27–30. doi: 10.1093/nar/28.1.27
|
| [14] |
Nakamura T, Yamada K D, Tomii K, et al. 2018. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics, 34(14): 2490–2492. doi: 10.1093/bioinformatics/bty121
|
| [15] |
Neilson J A D, Durnford D G. 2010. Structural and functional diversification of the light-harvesting complexes in photosynthetic eukaryotes. Photosynthesis Research, 106: 57–71. doi: 10.1007/s11120-010-9576-2
|
| [16] |
Niinemets Ü, Berry J A, von Caemmerer S, et al. 2017. Photosynthesis: ancient, essential, complex, diverse.. and in need of improvement in a changing world. New Phytologist, 213(1): 43–47. doi: 10.1111/nph.14307
|
| [17] |
Noctor G, Arisi A C M, Jouanin L, et al. 1999. Photorespiratory glycine enhances glutathione accumulation in both the chloroplastic and cytosolic compartments. Journal of Experimental Botany, 50(336): 1157–1167. doi: 10.1093/jxb/50.336.1157
|
| [18] |
R Development Core Team. 2014. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing
|
| [19] |
Smetacek V, Zingone A. 2013. Green and golden seaweed tides on the rise. Nature, 504(7478): 84–88. doi: 10.1038/nature12860
|
| [20] |
Suyama M, Torrents D, Bork P. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Research, 34(S2): W609–W612
|
| [21] |
Swift D G, Dunning L T, Igea J, et al. 2016. Evidence of positive selection associated with placental loss in tiger sharks. BMC Evolutionary Biology, 16(1): 126. doi: 10.1186/s12862-016-0696-y
|
| [22] |
Teng Linhong, Fan Xiao, Xu Dong, et al. 2017. Identification of genes under positive selection reveals differences in evolutionary adaptation between brown-algal species. Frontiers in Plant Science, 8: 1429. doi: 10.3389/fpls.2017.01429
|
| [23] |
Van Alstyne K L. 2008. Ecological and physiological roles of dimethylsulfoniopropionate and its products in marine macroalgae. In: Amsler C D, ed. Algal Chemical Ecology. Berlin, Heidelberg: Springer, 173–194
|
| [24] |
Vesty E F, Kessler R W, Wichard T, et al. 2015. Regulation of gametogenesis and zoosporogenesis in Ulva linza (Chlorophyta): comparison with Ulva mutabilis and potential for laboratory culture. Frontiers in Plant Science, 6: 15
|
| [25] |
Wichard T, Charrier B, Mineur F, et al. 2015. The green seaweed Ulva: a model system to study morphogenesis. Frontiers in Plant Science, 6: 72
|
| [26] |
Wingler A, Lea P J, Quick W P, et al. 2000. Photorespiration: metabolic pathways and their role in stress protection. Philosophical Transactions of the Royal Society B: Biological Sciences, 355(1402): 1517–1529. doi: 10.1098/rstb.2000.0712
|
| [27] |
Xie Chen, Mao Xizeng, Huang Jiaju, et al. 2011. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research, 39(S2): W316–W322
|
| [28] |
Xie Xiujun, Gu Wenhui, Gao Shan, et al. 2013. Alternative electron transports participate in the maintenance of violaxanthin de-epoxidase activity of Ulva sp. under low irradiance. PLoS One, 8(11): e78211
|
| [29] |
Yang Ziheng. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24(8): 1586–1591. doi: 10.1093/molbev/msm088
|
| [30] |
Yang Ziheng, Reis M D. 2011. Statistical properties of the branch-site test of positive selection. Molecular Biology and Evolution, 28(3): 1217–1228. doi: 10.1093/molbev/msq303
|
| [31] |
Zhang Junxiang, Yuan Hui, Yang Yong, et al. 2016. Plastid ribosomal protein S5 is involved in photosynthesis, plant development, and cold stress tolerance in Arabidopsis. Journal of Experimental Botany, 67(9): 2731–2744. doi: 10.1093/jxb/erw106
|
| [32] |
Zhang Xiaowen, Mou Shanli, Cao Shaona, et al. 2015. Roles of the transthylakoid proton gradient and xanthophyll cycle in the non-photochemical quenching of the green alga Ulva linza. Estuarine, Coastal and Shelf Science, 163: 69–74. doi: 10.1016/j.ecss.2014.09.006
|
| [33] |
Zhang Yongyu, He Peimin, Li Hongmei, et al. 2019. Ulva prolifera green-tide outbreaks and their environmental impact in the Yellow Sea, China. National Science Review, 6(4): 825–838. doi: 10.1093/nsr/nwz026
|
| 1. | Shabir A Rather, Kaikai Wang, Ting Wang, et al. Comparative chloroplast genome analysis reveals powerful barcodes for combatting illegal logging of CITES-listed threatened Asian rosewoods (Dalbergia, Leguminosae, Papilionoideae). Botanical Journal of the Linnean Society, 2024. doi:10.1093/botlinnean/boae086 |

Figures(2) / Tables(3)

Supported by:
Beijing Renhe Information Technology Co. Ltd

Jian Zhang, Xiaowen Zhang, Wentao Han, Xiao Fan, Yitao Wang, Dong Xu, Yan Zhang, Jian Ma, Chengwei Liang, Naihao Ye. Identification of genes under positive selection reveals evolutionary adaptation of Ulva mutabilis[J]. Acta Oceanologica Sinica, 2020, 39(10): 35-41. doi: 10.1007/s13131-020-1658-1

| Comparison | U. mutabilis | C. reinhardtii | V. carteri |
| Branch model | |||
| Free-radio model | |||
| Mean ω | 0.378 | 0.127 | 0.161 |
| Two-radio model | |||
| Number of GUPS (1<ω<3) | 9 | ||
| Site model | |||
| Number of GUPS | 242 | ||
| Number of GUPS (FDR<0.05) | 237 | ||
| Number of GUPS (PP>0.9) | 30 | ||
| Branch-site model | |||
| Number of GUPS | 67 | ||
| Number of GUPS (FDR<0.05) | 63 | ||
| Number of GUPS (PP>0.9) | 46 |
DownLoad:
CSV
| Protein ID | χ2 | p-value | Nr | KEGG |
| UM051_0030.1 | 3.649032 | 0.049775 | photosystem I reaction center subunit VI-chloroplastic-like | photosynthesis |
| UM041_0034.1 | 3.866748 | 0.049252 | 20S proteasome beta subunit | proteasome |
| UM025_0090.1 | 3.875197 | 0.049005 | transcription factor Tfb4 | basal transcription factors |
| UM020_0175.1 | 3.8908 | 0.048551 | cyclophilin-like protein | |
| UM017_0023.1 | 3.90656 | 0.048098 | type I inositol polyphosphate 5-phosphatase 1-like isoform X1 | |
| UM061_0055.1 | 3.92971 | 0.04744 | spermatogenesis-associated protein 4 | |
| UM020_0022.1 | 3.943346 | 0.047057 | MATE efflux family | |
| UM011_0045.1 | 3.952812 | 0.046793 | adenosine/AMP deaminase family protein | metabolic pathways |
| UM015_0094.1 | 3.96594 | 0.04643 | SET domain-containing protein | |
| UM002_0196.1 | 3.986382 | 0.045869 | tetratricopeptide repeat protein | |
| UM059_0039.1 | 3.988128 | 0.045822 | SET domain-containing protein | |
| UM005_0194.1 | 4.029512 | 0.044711 | metallo-hydrolase oxidoreductase | |
| UM119_0020.1 | 4.035582 | 0.04455 | dynein light chain, type 1 | |
| UM110_0011.1 | 4.076886 | 0.043474 | riboflavin biosynthesis chloroplastic | riboflavin metabolism |
| UM009_0042.1 | 4.089456 | 0.043152 | flavo protein | |
| UM119_0008.1 | 4.096726 | 0.042966 | indole-3-glycerol-phosphate synthase | |
| UM001_0277.1 | 4.121502 | 0.042341 | la-related protein 1A-like | |
| UM003_0103.1 | 4.175942 | 0.041002 | chlorophyll a-b binding protein of LHCII | photosynthesis-antenna protein |
| UM075_0040.1 | 4.193048 | 0.04059 | Sac domain-containing phosphoinositide phosphatase | |
| UM020_0063.1 | 4.271742 | 0.038751 | 50S ribosomal protein L3-1, chloroplastic | ribosome |
| UM101_0006.1 | 4.318366 | 0.037703 | tubulin-tyrosine ligase | |
| UM002_0307.1 | 4.357032 | 0.036856 | nucleotide-diphospho-sugar transferase domain | |
| UM033_0058.1 | 4.399316 | 0.035953 | vacuolar fusion protein MON1 homolog isoform X2 | |
| UM022_0088.1 | 4.428682 | 0.03534 | DUF455 family | |
| UM004_0158.1 | 4.448474 | 0.034932 | integral membrane protein TerC, riboswitch-linked | |
| UM001_0470.1 | 4.459038 | 0.034717 | WD repeat-containing protein 6 isoform X1 | |
| UM004_0249.1 | 4.51936 | 0.033513 | ABC transporter F family member-like | |
| UM095_0035.1 | 4.519716 | 0.033506 | ubiquinone biosynthesis protein, partial | |
| UM019_0140.1 | 4.552578 | 0.032869 | 40S ribosomal protein S8 | ribosome |
| UM100_0037.1 | 4.616152 | 0.031672 | hypothetical protein | |
| UM133_0013.1 | 4.619028 | 0.031619 | Reticulata-related chloroplastic-like | |
| UM077_0057.1 | 4.643308 | 0.031175 | IMPACT isoform X1 | |
| UM005_0088.1 | 4.64428 | 0.031157 | thylakoid lumenal protein | |
| UM005_0011.1 | 4.647732 | 0.031095 | RNA polymerase II-associated factor 1-like protein | |
| UM007_0229.1 | 4.671324 | 0.03067 | sorting nexin 2a | |
| UM035_0106.1 | 4.861868 | 0.027457 | ribosome 60S biogenesis N-terminal-domain-containing protein | |
| UM098_0047.1 | 4.880034 | 0.027169 | phospholipase A I-like isoform X2 | |
| UM040_0040.1 | 4.933432 | 0.026342 | CUE domain-containing protein | |
| UM057_0023.1 | 5.000268 | 0.025343 | COMPASS-like H3K4 histone methylase component WDR5A | |
| UM085_0051.1 | 5.080884 | 0.024191 | GPI inositol-deacylase PGAP1-like isoform B | |
| UM062_0033.1 | 5.242224 | 0.022045 | argininosuccinate synthase | alanine, aspartate metabolism |
| UM014_0155.1 | 5.399928 | 0.020138 | polysulfide reductase | |
| UM066_0033.1 | 5.46381 | 0.019414 | epsilon-COP | |
| UM020_0154.1 | 5.538692 | 0.0186 | transcription factor bHLH34 | |
| UM066_0060.1 | 5.542258 | 0.018563 | L-isoaspartate(D-aspartate) O-methyltransferase | |
| UM041_0094.1 | 5.61881 | 0.017769 | prephenate dehydratase | biosynthesis of amino acids |
| UM051_0040.1 | 5.620024 | 0.017756 | centrosomal protein of 78 kDa | |
| UM047_0010.1 | 5.762886 | 0.016368 | phosphatidate phosphatase PAH1 isoform X1 | glycerophospholipid metabolism |
| UM002_0428.1 | 5.880034 | 0.015314 | PREDICTED: nuclear-interacting partner of ALK isoform X1 | |
| UM020_0144.1 | 6.001238 | 0.014296 | kinesin light chain 3 isoform X1 | |
| UM028_0126.1 | 6.071472 | 0.013738 | glycine cleavage system H protein, mitochondrial | glycine, serine and threonine metabolism |
| UM023_0033.1 | 6.119312 | 0.013371 | adenylyl cyclase class-3/4/guanylyl cyclase | purine metabolism |
| to be continued | ||||
DownLoad:
CSV
| Model | Gene ID | Function description in Nr and Pfam database | Gene ID | Function description in Nr and Pfam database |
| Branch-model | UM011_0230.1 | p25-alpha | UM068_0038.1 | polyketide cyclase/dehydrase and lipid transport |
| UM011_0231.1 | UM094_0042.1 | |||
| Site-model | UM031_0027.1 | glucokinase | UM005_0209.1 | aminotransferase class I and II |
| UM031_0028.1 | UM005_0214.1 | |||
| UM146_0032.1 | N2, N2-dimethylguanosine tRNA methyltranse | UM008_0174.1 | hypothetical protein | |
| UM146_0033.1 | UM281_0004.1 | |||
| UM013_0057.1 | Dor1-like family | UM100_0006.1 | chloroplastic isoform | |
| UM060_0118.1 | UM100_0007.1 | |||
| UM049_0058.1 | ubiquitin-conjugating enzyme | UM018_0136.1 | peptidase family M41 | |
| UM058_0003.1 | UM020_0045.1 | |||
| UM012_0017.1 | protein kinase domain | UM015_0045.1 | protein tyrosine kinase | |
| UM149_0036.1 | UM015_0046.1 | |||
| UM093_0008.1 | recA bacterial DNA recombination protein | UM001_0125.1 | ubiquitin-specific protease | |
| UM093_0026.1 | UM001_0129.1 | |||
| UM005_0351.1 | cytochrome C biogenesis protein | UM001_0491.1 | FAD dependent oxidoreductase | |
| UM077_0032.1 | UM001_0492.1 | |||
| UM010_0035.1 | TCP-1/cpn60 chaperonin | UM002_0402.1 | DNL zinc finger | |
| UM011_0180.1 | UM018_0127.1 | |||
| UM011_0077.1 | hypothetical protein | UM037_0017.1 | protein of unknown function (DUF3250) | |
| UM011_0096.1 | UM037_0018.1 | |||
| UM037_0056.1 | no hit | UM034_0001.1 | FKBP-type peptidyl-prolyl cis-trans isomerase | |
| UM044_0086.1 | UM034_0003.1 | |||
| UM009_0050.1 | CobW/HypB/UreG, nucleotide-binding domain | UM008_0173.1 | WD domain, G-beta repeat | |
| UM092_0039.1 | UM281_0003.1 | |||
| UM069_0030.1 | carbamoyl-phosphate synthase small chain, CPSase domain | UM015_0020.1 | cation efflux family | |
| UM309_0004.1 | UM026_0097.1 | |||
| UM043_0048.1 | aminotransferase class I and II | UM012_0077.1 | no hit | |
| UM057_0008.1 | UM131_0006.1 | |||
| UM001_0588.1 | plasma-membrane choline transporter | UM001_0573.1 | Sec63 Brl domain | |
| UM001_0591.1 | UM002_0245.1 | |||
| UM035_0019.1 | TIP41-like family | UM003_0004.1 | ATP12 chaperone protein | |
| UM035_0020.1 | UM047_0029.1 | |||
| UM010_0149.1 | TspO/MBR | UM007_0020.1 | ABC transporter | |
| UM010_0150.1 | UM139_0019.1 | |||
| UM012_0035.1 | Hsp70 protein | UM008_0176.1 | enoyl-(acyl carrier protein) reductase | |
| UM012_0036.1 | UM281_0001.1 | |||
| UM018_0002.1 | RNA methyltransferase | UM103_0009.1 | no hit | |
| to be continued | ||||
DownLoad:
CSV
| Comparison | U. mutabilis | C. reinhardtii | V. carteri |
| Branch model | |||
| Free-radio model | |||
| Mean ω | 0.378 | 0.127 | 0.161 |
| Two-radio model | |||
| Number of GUPS (1<ω<3) | 9 | ||
| Site model | |||
| Number of GUPS | 242 | ||
| Number of GUPS (FDR<0.05) | 237 | ||
| Number of GUPS (PP>0.9) | 30 | ||
| Branch-site model | |||
| Number of GUPS | 67 | ||
| Number of GUPS (FDR<0.05) | 63 | ||
| Number of GUPS (PP>0.9) | 46 |
| Protein ID | χ2 | p-value | Nr | KEGG |
| UM051_0030.1 | 3.649032 | 0.049775 | photosystem I reaction center subunit VI-chloroplastic-like | photosynthesis |
| UM041_0034.1 | 3.866748 | 0.049252 | 20S proteasome beta subunit | proteasome |
| UM025_0090.1 | 3.875197 | 0.049005 | transcription factor Tfb4 | basal transcription factors |
| UM020_0175.1 | 3.8908 | 0.048551 | cyclophilin-like protein | |
| UM017_0023.1 | 3.90656 | 0.048098 | type I inositol polyphosphate 5-phosphatase 1-like isoform X1 | |
| UM061_0055.1 | 3.92971 | 0.04744 | spermatogenesis-associated protein 4 | |
| UM020_0022.1 | 3.943346 | 0.047057 | MATE efflux family | |
| UM011_0045.1 | 3.952812 | 0.046793 | adenosine/AMP deaminase family protein | metabolic pathways |
| UM015_0094.1 | 3.96594 | 0.04643 | SET domain-containing protein | |
| UM002_0196.1 | 3.986382 | 0.045869 | tetratricopeptide repeat protein | |
| UM059_0039.1 | 3.988128 | 0.045822 | SET domain-containing protein | |
| UM005_0194.1 | 4.029512 | 0.044711 | metallo-hydrolase oxidoreductase | |
| UM119_0020.1 | 4.035582 | 0.04455 | dynein light chain, type 1 | |
| UM110_0011.1 | 4.076886 | 0.043474 | riboflavin biosynthesis chloroplastic | riboflavin metabolism |
| UM009_0042.1 | 4.089456 | 0.043152 | flavo protein | |
| UM119_0008.1 | 4.096726 | 0.042966 | indole-3-glycerol-phosphate synthase | |
| UM001_0277.1 | 4.121502 | 0.042341 | la-related protein 1A-like | |
| UM003_0103.1 | 4.175942 | 0.041002 | chlorophyll a-b binding protein of LHCII | photosynthesis-antenna protein |
| UM075_0040.1 | 4.193048 | 0.04059 | Sac domain-containing phosphoinositide phosphatase | |
| UM020_0063.1 | 4.271742 | 0.038751 | 50S ribosomal protein L3-1, chloroplastic | ribosome |
| UM101_0006.1 | 4.318366 | 0.037703 | tubulin-tyrosine ligase | |
| UM002_0307.1 | 4.357032 | 0.036856 | nucleotide-diphospho-sugar transferase domain | |
| UM033_0058.1 | 4.399316 | 0.035953 | vacuolar fusion protein MON1 homolog isoform X2 | |
| UM022_0088.1 | 4.428682 | 0.03534 | DUF455 family | |
| UM004_0158.1 | 4.448474 | 0.034932 | integral membrane protein TerC, riboswitch-linked | |
| UM001_0470.1 | 4.459038 | 0.034717 | WD repeat-containing protein 6 isoform X1 | |
| UM004_0249.1 | 4.51936 | 0.033513 | ABC transporter F family member-like | |
| UM095_0035.1 | 4.519716 | 0.033506 | ubiquinone biosynthesis protein, partial | |
| UM019_0140.1 | 4.552578 | 0.032869 | 40S ribosomal protein S8 | ribosome |
| UM100_0037.1 | 4.616152 | 0.031672 | hypothetical protein | |
| UM133_0013.1 | 4.619028 | 0.031619 | Reticulata-related chloroplastic-like | |
| UM077_0057.1 | 4.643308 | 0.031175 | IMPACT isoform X1 | |
| UM005_0088.1 | 4.64428 | 0.031157 | thylakoid lumenal protein | |
| UM005_0011.1 | 4.647732 | 0.031095 | RNA polymerase II-associated factor 1-like protein | |
| UM007_0229.1 | 4.671324 | 0.03067 | sorting nexin 2a | |
| UM035_0106.1 | 4.861868 | 0.027457 | ribosome 60S biogenesis N-terminal-domain-containing protein | |
| UM098_0047.1 | 4.880034 | 0.027169 | phospholipase A I-like isoform X2 | |
| UM040_0040.1 | 4.933432 | 0.026342 | CUE domain-containing protein | |
| UM057_0023.1 | 5.000268 | 0.025343 | COMPASS-like H3K4 histone methylase component WDR5A | |
| UM085_0051.1 | 5.080884 | 0.024191 | GPI inositol-deacylase PGAP1-like isoform B | |
| UM062_0033.1 | 5.242224 | 0.022045 | argininosuccinate synthase | alanine, aspartate metabolism |
| UM014_0155.1 | 5.399928 | 0.020138 | polysulfide reductase | |
| UM066_0033.1 | 5.46381 | 0.019414 | epsilon-COP | |
| UM020_0154.1 | 5.538692 | 0.0186 | transcription factor bHLH34 | |
| UM066_0060.1 | 5.542258 | 0.018563 | L-isoaspartate(D-aspartate) O-methyltransferase | |
| UM041_0094.1 | 5.61881 | 0.017769 | prephenate dehydratase | biosynthesis of amino acids |
| UM051_0040.1 | 5.620024 | 0.017756 | centrosomal protein of 78 kDa | |
| UM047_0010.1 | 5.762886 | 0.016368 | phosphatidate phosphatase PAH1 isoform X1 | glycerophospholipid metabolism |
| UM002_0428.1 | 5.880034 | 0.015314 | PREDICTED: nuclear-interacting partner of ALK isoform X1 | |
| UM020_0144.1 | 6.001238 | 0.014296 | kinesin light chain 3 isoform X1 | |
| UM028_0126.1 | 6.071472 | 0.013738 | glycine cleavage system H protein, mitochondrial | glycine, serine and threonine metabolism |
| UM023_0033.1 | 6.119312 | 0.013371 | adenylyl cyclase class-3/4/guanylyl cyclase | purine metabolism |
| to be continued | ||||
| Model | Gene ID | Function description in Nr and Pfam database | Gene ID | Function description in Nr and Pfam database |
| Branch-model | UM011_0230.1 | p25-alpha | UM068_0038.1 | polyketide cyclase/dehydrase and lipid transport |
| UM011_0231.1 | UM094_0042.1 | |||
| Site-model | UM031_0027.1 | glucokinase | UM005_0209.1 | aminotransferase class I and II |
| UM031_0028.1 | UM005_0214.1 | |||
| UM146_0032.1 | N2, N2-dimethylguanosine tRNA methyltranse | UM008_0174.1 | hypothetical protein | |
| UM146_0033.1 | UM281_0004.1 | |||
| UM013_0057.1 | Dor1-like family | UM100_0006.1 | chloroplastic isoform | |
| UM060_0118.1 | UM100_0007.1 | |||
| UM049_0058.1 | ubiquitin-conjugating enzyme | UM018_0136.1 | peptidase family M41 | |
| UM058_0003.1 | UM020_0045.1 | |||
| UM012_0017.1 | protein kinase domain | UM015_0045.1 | protein tyrosine kinase | |
| UM149_0036.1 | UM015_0046.1 | |||
| UM093_0008.1 | recA bacterial DNA recombination protein | UM001_0125.1 | ubiquitin-specific protease | |
| UM093_0026.1 | UM001_0129.1 | |||
| UM005_0351.1 | cytochrome C biogenesis protein | UM001_0491.1 | FAD dependent oxidoreductase | |
| UM077_0032.1 | UM001_0492.1 | |||
| UM010_0035.1 | TCP-1/cpn60 chaperonin | UM002_0402.1 | DNL zinc finger | |
| UM011_0180.1 | UM018_0127.1 | |||
| UM011_0077.1 | hypothetical protein | UM037_0017.1 | protein of unknown function (DUF3250) | |
| UM011_0096.1 | UM037_0018.1 | |||
| UM037_0056.1 | no hit | UM034_0001.1 | FKBP-type peptidyl-prolyl cis-trans isomerase | |
| UM044_0086.1 | UM034_0003.1 | |||
| UM009_0050.1 | CobW/HypB/UreG, nucleotide-binding domain | UM008_0173.1 | WD domain, G-beta repeat | |
| UM092_0039.1 | UM281_0003.1 | |||
| UM069_0030.1 | carbamoyl-phosphate synthase small chain, CPSase domain | UM015_0020.1 | cation efflux family | |
| UM309_0004.1 | UM026_0097.1 | |||
| UM043_0048.1 | aminotransferase class I and II | UM012_0077.1 | no hit | |
| UM057_0008.1 | UM131_0006.1 | |||
| UM001_0588.1 | plasma-membrane choline transporter | UM001_0573.1 | Sec63 Brl domain | |
| UM001_0591.1 | UM002_0245.1 | |||
| UM035_0019.1 | TIP41-like family | UM003_0004.1 | ATP12 chaperone protein | |
| UM035_0020.1 | UM047_0029.1 | |||
| UM010_0149.1 | TspO/MBR | UM007_0020.1 | ABC transporter | |
| UM010_0150.1 | UM139_0019.1 | |||
| UM012_0035.1 | Hsp70 protein | UM008_0176.1 | enoyl-(acyl carrier protein) reductase | |
| UM012_0036.1 | UM281_0001.1 | |||
| UM018_0002.1 | RNA methyltransferase | UM103_0009.1 | no hit | |
| to be continued | ||||
 DownLoad:
DownLoad:

 DownLoad:
DownLoad: